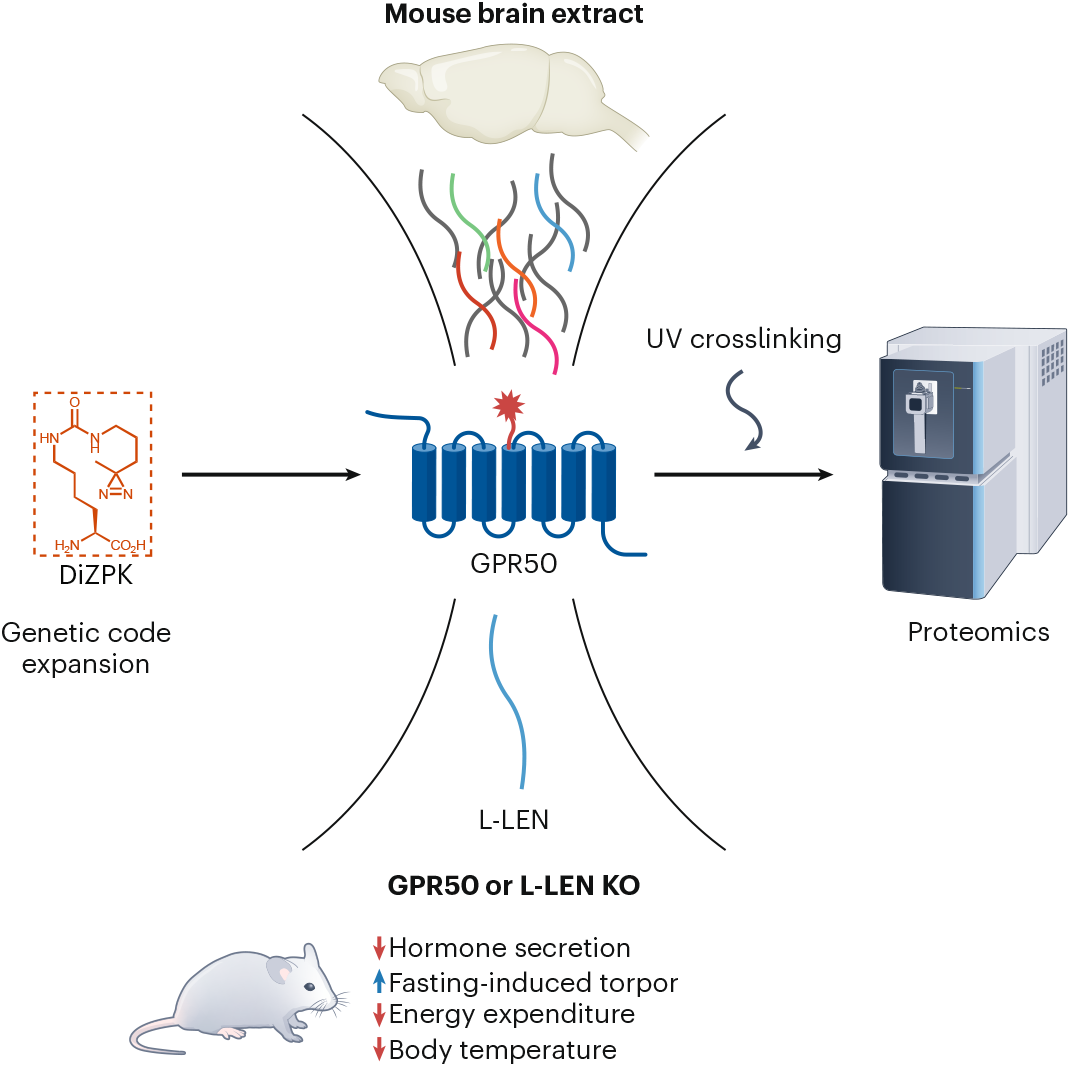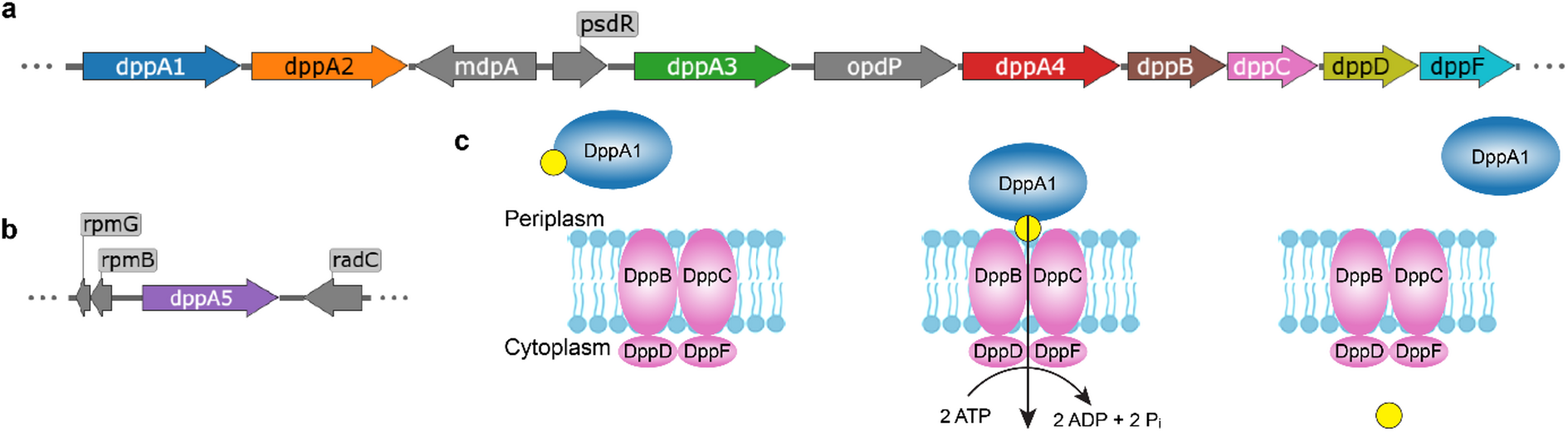
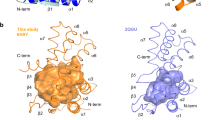


Pseudomonas aeruginosa is a notorious opportunistic pathogen, and part of its survival strategy is a remarkably flexible nutrient uptake network. One of the key systems it uses is the dipeptide permease, or Dpp, which imports short peptides from the periplasm into the cell. What makes this system unusual is that, unlike the single solute-binding protein seen in many bacteria, P. aeruginosa encodes five DppA paralogs.
A new study has now mapped the binding preferences of all five proteins, revealing that they are far from interchangeable. Using differential scanning fluorimetry and a library of 281 di- and tripeptides, the researchers found clear specialization among the paralogs. DppA1 and DppA3 showed a stronger preference for dipeptides, while DppA2 and DppA4 were more responsive to tripeptides. DppA5, by contrast, showed no detectable peptide binding under the conditions tested.
The results suggest that the multiple DppA proteins do not simply duplicate one another’s function. Instead, they appear to broaden the range of peptide substrates that P. aeruginosa can capture from its environment. That flexibility may help the bacterium thrive in nutrient-poor or competitive settings, including the human host.
The work also reinforces the idea that peptide transport in this pathogen may be tied to more than metabolism alone. Dpp-related proteins have previously been linked to behaviors such as biofilm formation and chemotaxis, raising the possibility that ligand recognition by DppA proteins could influence signaling as well as uptake.
From a practical standpoint, the study offers two important takeaways. First, it corrects the assumption that the five paralogs are functionally redundant. Second, it provides a more detailed foundation for thinking about Dpp as a potential route for peptide-based drug delivery strategies, including Trojan horse antimicrobials designed to exploit bacterial transport systems.
In short, P. aeruginosa appears to use a diversified peptide-sensing toolkit to expand what it can import, and that diversification may be part of what makes this pathogen such a resilient problem in clinical settings.