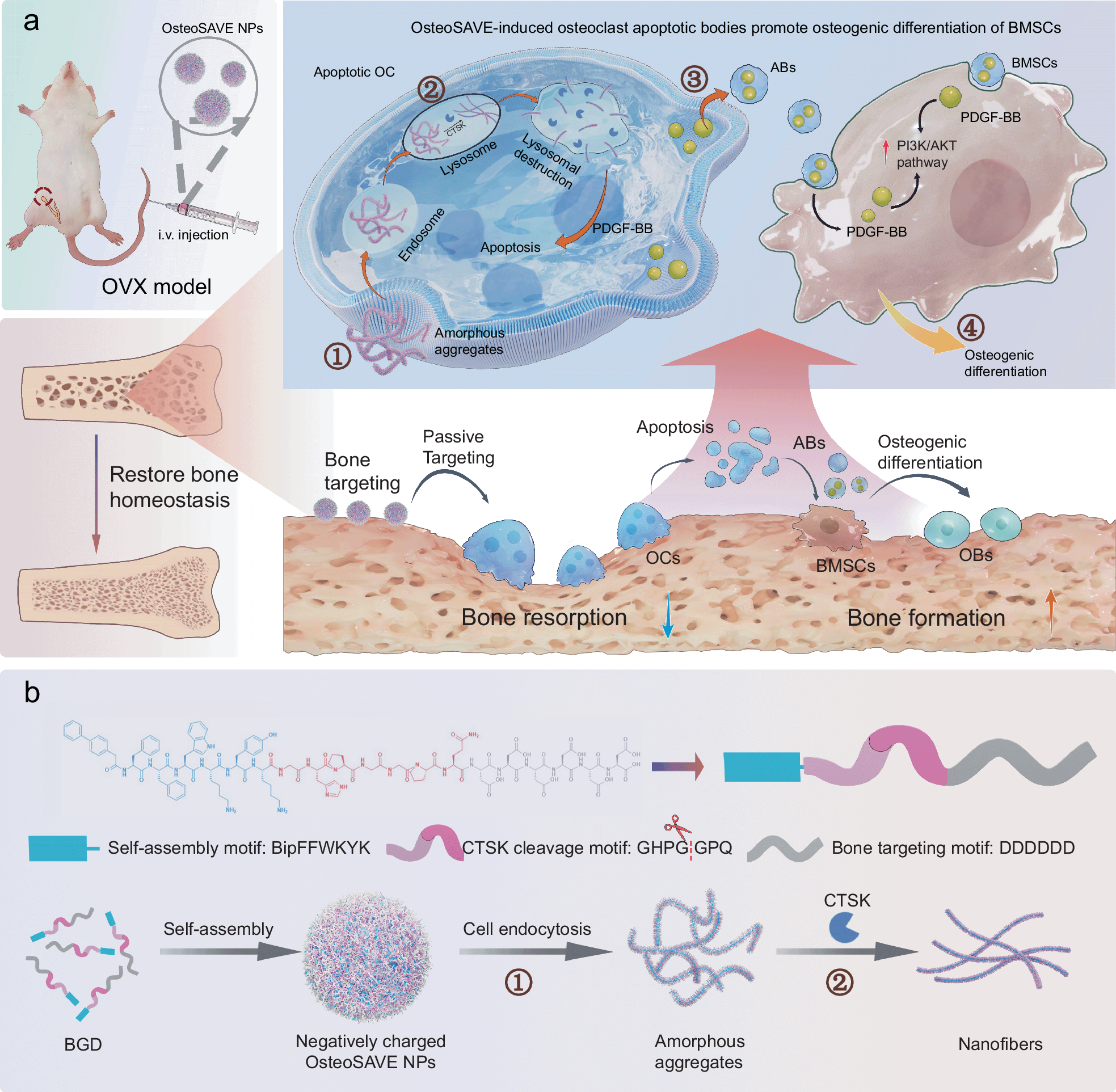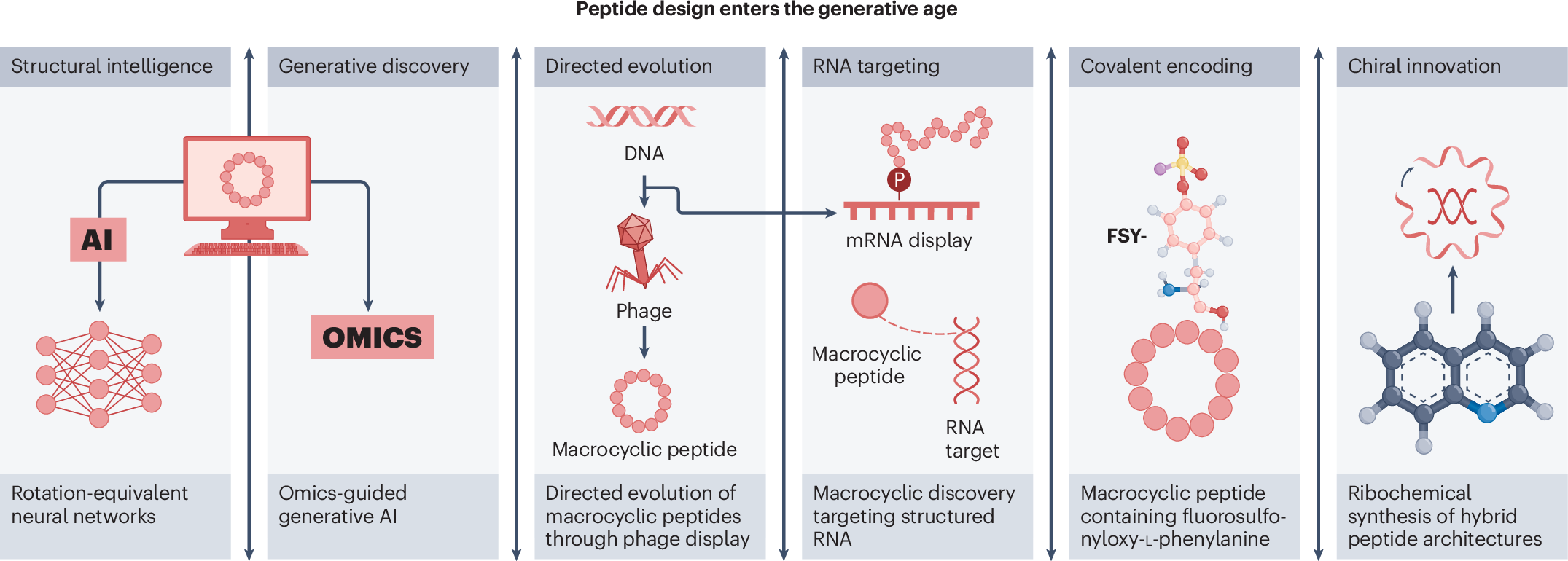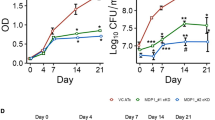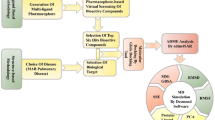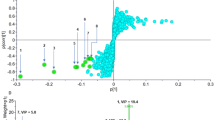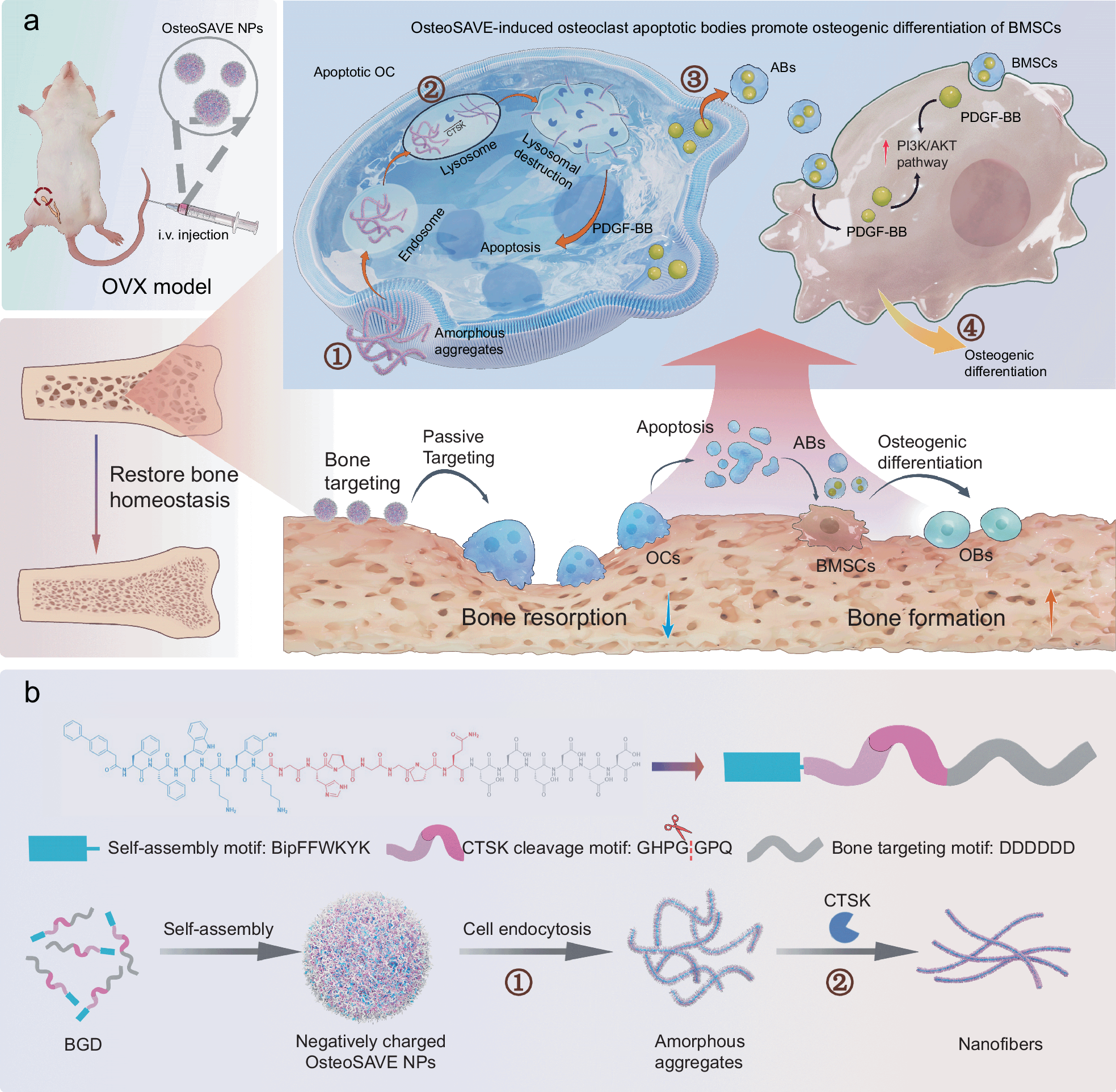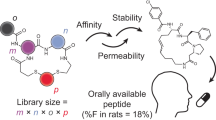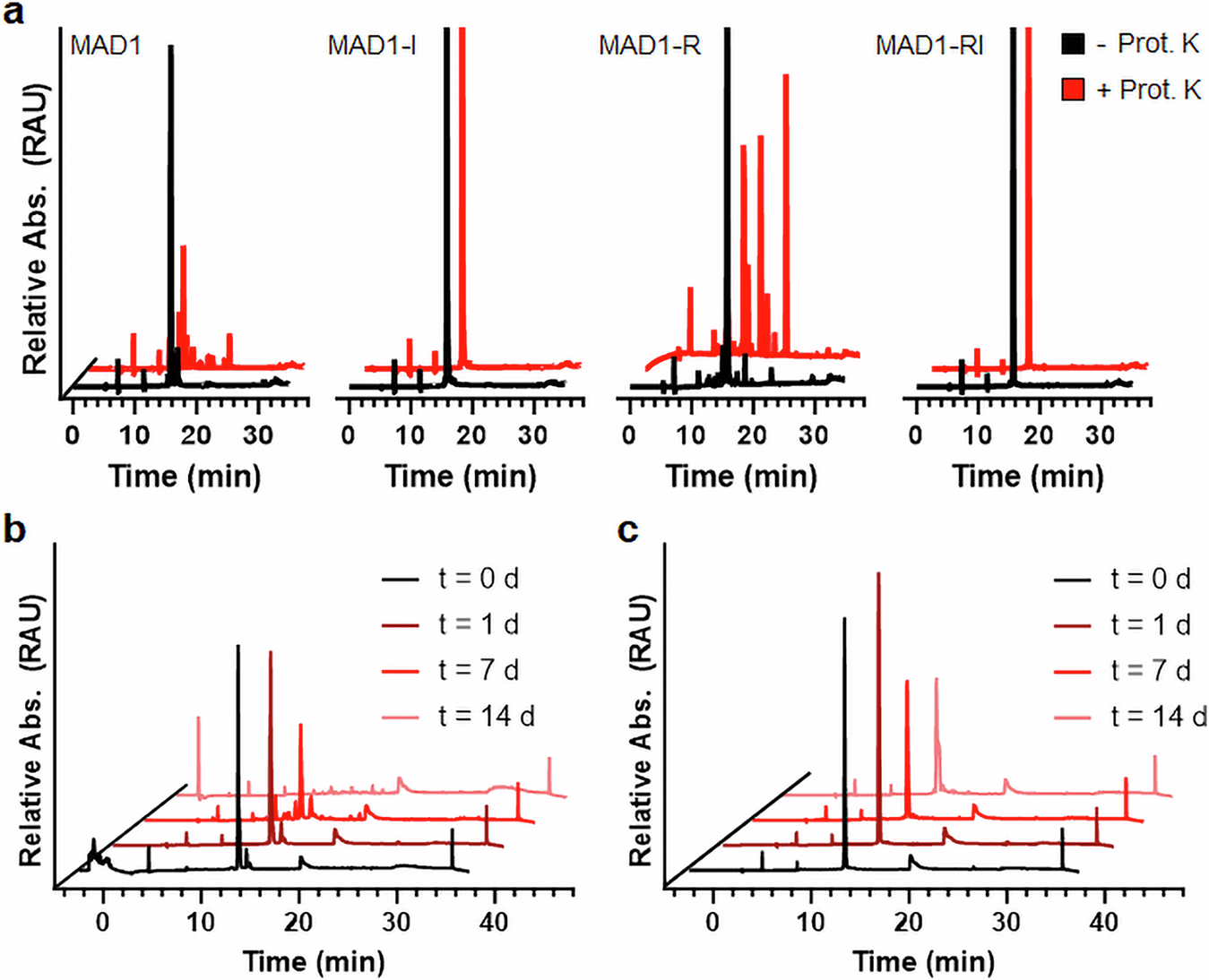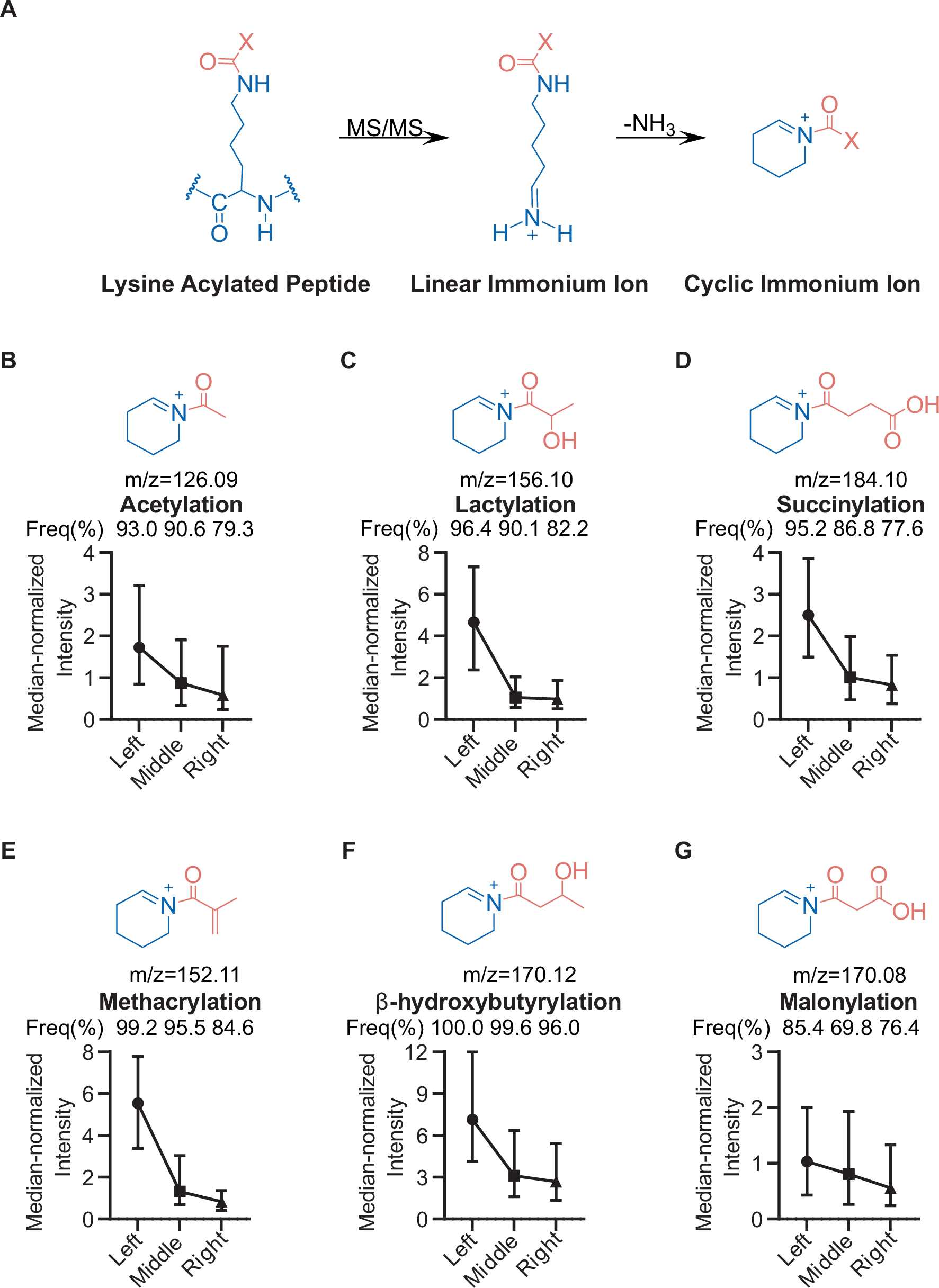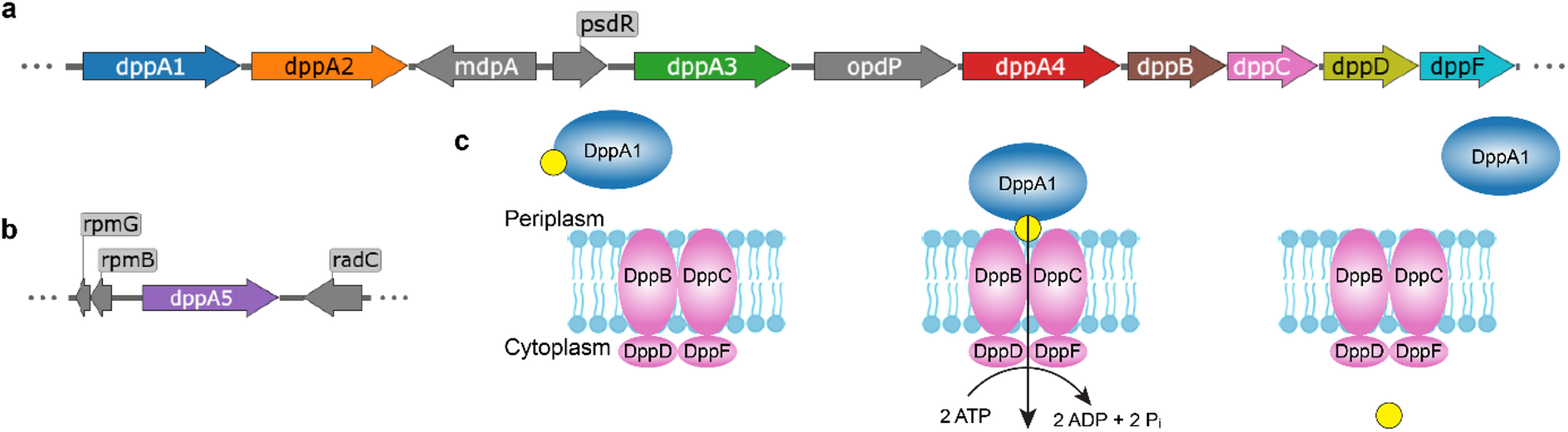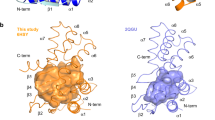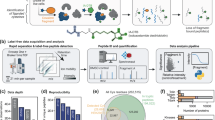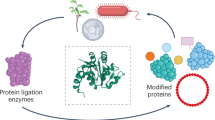
Peptide chemistry is entering a new phase. Instead of focusing mainly on making more analogues of known sequences, researchers are now combining synthesis, display technologies, and computational design to build peptides with increasingly precise functions.
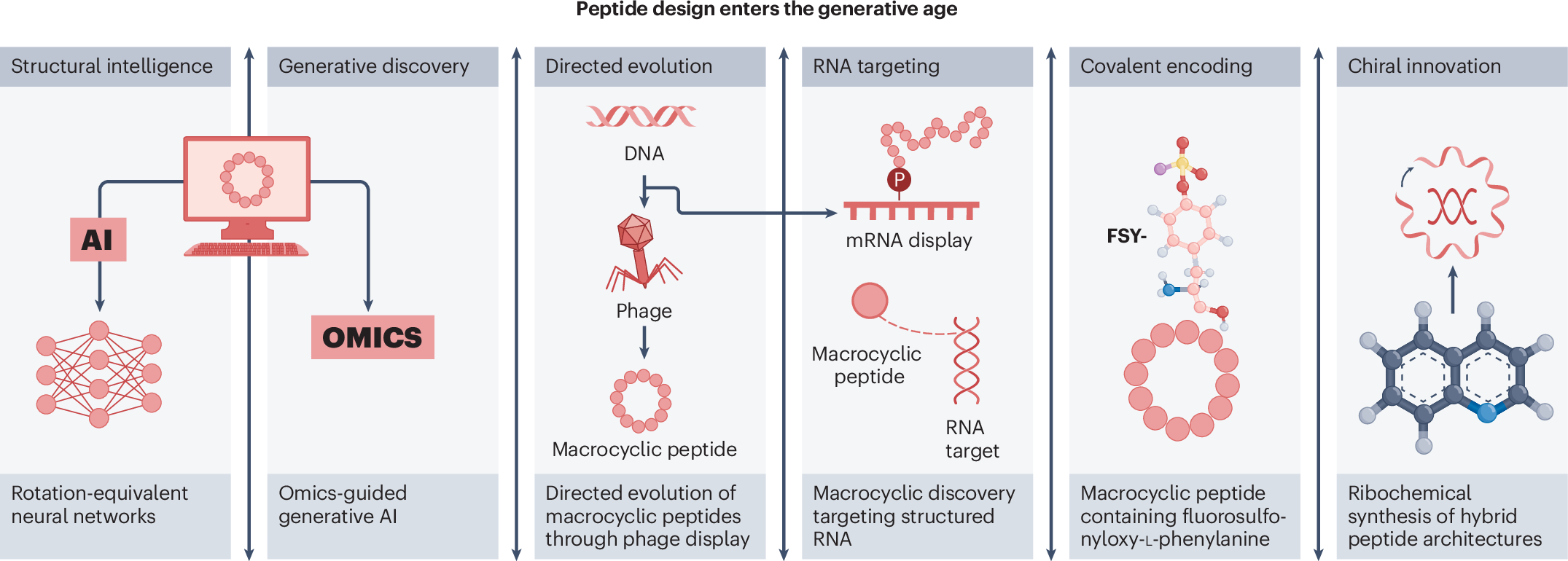
A recent review of the field points to a clear shift in 2025: peptides are being treated less like static chains of amino acids and more like programmable molecular systems. Artificial intelligence is helping researchers predict and optimize binders, while new synthetic methods are making it easier to explore macrocycles, constrained shapes, and covalent interactions.
Why this matters
Traditional peptide discovery often relied on screening large numbers of variants and then refining the best hits. That approach still matters, but recent progress suggests a broader toolkit is now available. By using deep learning and improved molecular modelling, scientists can design peptide structures with much higher structural specificity before they are ever synthesized.
This is especially important for targets that have been difficult to address with conventional small molecules or linear peptides. Macrocyclic architectures, for example, can improve binding affinity, selectivity, and resistance to degradation. They also create new possibilities for engaging challenging biomolecules such as proteins and structured RNA.
Examples from the 2025 literature
The studies highlighted alongside the review show how diverse the field has become:
- Deep learning has been used to design high-affinity protein-binding macrocycles de novo.
- Computational venom mining has been applied to antimicrobial discovery.
- Macrocyclic phage display has been used to identify selective protease substrates.
- mRNA display has enabled discovery of macrocyclic peptide binders, covalent modifiers, and degraders of structured RNA.
- Ribosomal methods have incorporated unusual amino acids to create target-specific covalent binders.
- Hybrid chemical and ribosomal synthesis has expanded access to atropisomeric and macrocyclic peptides with embedded quinoline motifs.
From binders to functional molecules
The broader message is that peptide research is no longer limited to finding molecules that simply stick to a target. The new goal is to engineer function. In practice, that means designing peptides that can bind, react, degrade, or otherwise modulate biological systems in a controlled way.
Macrocyclic and covalent frameworks are particularly valuable here because they add chemical constraints that improve performance and increase the range of possible mechanisms. When paired with AI-guided design, these frameworks can make peptide discovery faster, more selective, and more inventive.
The outlook
As synthesis and computation continue to merge, peptide chemistry is becoming more modular and predictive. The field appears to be moving toward a future in which peptide sequences are not just screened, but designed as tailored molecular devices for specific tasks in biology and medicine.
For peptide researchers, that shift is significant: the opportunity is no longer only to accumulate analogues, but to build entirely new classes of functional peptide architectures.