
Host-defense peptides have long been viewed as attractive antibiotic candidates because they attack microbes in ways that differ from classic small-molecule drugs. Their main weakness is stability: natural peptides are quickly broken down by proteases, so researchers often turn to retro-inversion, a design strategy that flips both the peptide backbone direction and amino acid chirality.
That approach usually improves durability, but it can also reduce activity by changing folding and self-assembly. In a new report, researchers show that the opposite can happen for antimycobacterial peptides. For peptides aimed at mycobacteria, retro-inversion increased potency and improved selectivity without broadly increasing toxicity toward non-target species.
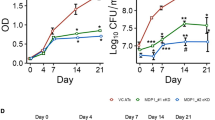
The strongest example in the study was a retro-inverted version of a lead peptide called MAD1, named MAD1-RI. In laboratory tests, it rapidly sterilized replicating Mycobacterium tuberculosis cultures, worked against drug-resistant clinical isolates, and boosted the effect of co-administered TB antibiotics. The authors also found signs that the mechanism was not simply about protease resistance; instead, the altered peptide architecture appeared to change how it interacted with the mycobacterial membrane.
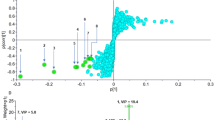
Transcriptomic and biophysical analyses supported a model in which MAD1-RI triggers membrane stress along with downstream metabolic disruption. Taken together, the findings suggest retro-inversion may be a useful, and somewhat unexpected, design tool for building non-natural peptides with stronger antimycobacterial activity.
The work is especially notable given the growing need for new treatments against both tuberculosis and non-tuberculous mycobacterial infections, including strains with broad drug resistance. If these results translate beyond the lab, retro-inverted peptides could become a promising new class of targeted anti-mycobacterial agents.





{kind=link}